Naproxeno
Tabletas, suspensión oral
Antirreumático y antiinflamatorio
no esteroideo
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Naproxeno....................................................................... 250 mg
Cada TABLETA contiene:
Naproxeno....................................................................... 500 mg
Cada 100 ml de SUSPENSIÓN contienen:
Naproxeno....................................................................... 125 mg
INDICACIONES TERAPÉUTICAS:
NAPROXENO es un miembro del grupo ácido arilacético de fármacos antiinflamatorios no esteroideos.
NAPROXENO en forma de tabletas convencionales está indicado para el tratamiento de la artritis reumatoide, osteoartritis, espondilitis anquilosante y artritis juvenil. También está indicado para el tratamiento de tendinitis, bursitis, esguinces y para el manejo del dolor posquirúrgico.
CONTRAINDICACIONES:
NAPROXENO está contraindicado en pacientes que tienen reacciones alérgicas a la prescripción. También está contraindicado en pacientes en quienes el ácido acetilsalicílico u otros agentes analgésicos antiinflamatorios no esteroidales inducen el síndrome de asma, rinitis y pólipos nasales. Ambos tipos de reacciones pueden ser fatales.
Las reacciones anafilactoides al NAPROXENO, ya sean el tipo alérgico verdadero o la idiosincrasia farmacológica (por ejemplo, síndrome de hipersensibilidad al ácido acetilsalicílico), usualmente ocurren en pacientes con antecedentes conocidos a esas reacciones.
Por tanto, antes de empezar la terapia es importante investigar cuidadosamente con el paciente aspectos como asma, pólipos nasales, urticaria e hipotensión, asociados con fármacos antiinflamatorios no esteroidales. Además, el tratamiento debe ser suspendido si durante la terapia ocurren estos síntomas. No se ha establecido la seguridad y efectividad en niños menores de 2 años.
PRECAUCIONES GENERALES:
Se debe evitar el uso concomitante de otros productos que contengan naproxeno.
Riesgo de ulceraciones, sangrado y perforación gastrointestinal durante la terapia con agentes antiinflamatorios no esteroidales: En los pacientes tratados crónicamente con terapia AINE en cualquier momento puede ocurrir toxicidad gastrointestinal severa, como sangrado, ulceración y perforación, con o sin síntomas de advertencia. Aunque son comunes los problemas menores en el tracto gastrointestinal, como dispepsia, por lo usual desarrollándose de manera temprana durante la terapia, los médicos deben permanecer alertas en lo que respecta a ulceración y sangrado en los pacientes tratados crónicamente con AINE, aun en ausencia de síntomas previos del tracto gastrointestinal.
En pacientes observados en estudios clínicos de varios meses a dos años de duración, parecieron ocurrir úlceras sintomáticas en el tracto gastrointestinal superior, sangrado evidente o perforación en aproximadamente 1% de los pacientes tratados durante 3 a 6 meses, y en aproximadamente 2 a 4% de los pacientes tratados durante 1 año. Los médicos deben informar a los pacientes acerca de los signos y/o síntomas de toxicidad gastrointestinal severa y de las medidas que deben tomar si esto ocurre.
Los estudios realizados hasta la fecha con todos los productos de NAPROXENO no han identificado ningún subgrupo de pacientes que no estén en riesgo de desarrollar ulceración péptica y sangrado. No hay alguna diferencia entre los diferentes productos de NAPROXENO en su propensión a causar ulceración péptica y sangrado. No se conocen otros factores que aumenten el riesgo de sangrado, excepto por una historia previa de eventos gastrointestinales severos y otros factores de riesgo, conocidos como asociados con enfermedad péptica ulcerosa, alcoholismo, tabaquismo, etcétera. Los pacientes ancianos o debilitados parecen tolerar la ulceración o el sangrado más o menos que otros individuos, y la mayoría de los reportes espontáneos de eventos gastrointestinales fatales ocurren en esta población. Los estudios realizados hasta la fecha no son concluyentes en lo que se refiere al riesgo relativo de varios AINEs para causar esas reacciones.
Dosis altas de cualquier AINE probablemente conllevan un mayor riesgo de estas reacciones, aunque los estudios clínicos controlados muestran que esto no sucede en la mayoría de los casos.
Considerando el uso de dosis relativamente altas (dentro del intervalo de dosis recomendado), se debe anticipar un beneficio suficiente que supere el posible riesgo aumentado de toxicidad gastrointestinal.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
El anión NAPROXENO se ha encontrado en la leche en mujeres que amamantan, a una concentración aproximadamente de 1% de la encontrada en plasma. Dada la posibilidad de reacciones adversas de los fármacos inhibidores de la prostaglandina en los neonatos, se debe evitar el uso de NAPROXENO en mujeres que amamantan.
REACCIONES SECUNDARIAS Y ADVERSAS:
Los pacientes con valores iniciales de hemoglobina de 10 gramos o menores, que van a recibir terapia prolongada, deben someterse a determinaciones periódicas de los valores de hemoglobina.
Las actividades antipirética y antiinflamatoria del fármaco pueden disminuir la fiebre e inflamación, disminuyendo así su utilidad como signos diagnósticos para detectar complicaciones de otros cuadros.
Efectos renales: Al igual que con otros fármacos antiinflamatorios no esteroideos, la administración prolongada de NAPROXENO en animales ha resultado en necrosis papilar renal y otras patologías renales anormales.
En humanos se han presentado reportes de nefritis intersticial, hematuria, proteinuria, y ocasionalmente, síndrome nefrótico asociado con productos que contienen NAPROXENO y otros AINEs, desde su introducción en el mercado.
Se ha visto una segunda forma de toxicidad renal en pacientes que toman NAPROXENO, así como otros fármacos antiinflamatorios no esteroideos.
En pacientes con condiciones prerrenales que originan una reducción del flujo renal sanguíneo o del volumen sanguíneo, donde las prostaglandinas renales tienen una función de apoyo en el mantenimiento de la perfusión renal, la administración de un fármaco antiinflamatorio no esteroideo puede causar una disminución dependiente de la dosis en la formación de prostaglandina, y precipitar descompensación renal franca. Los pacientes en mayor riesgo de esta reacción son aquéllos con insuficiencia renal, insuficiencia cardiaca, trastornos hepáticos, pacientes que toman diuréticos y los ancianos.
La interrupción de la terapia con agentes antiinflamatorios no esteroideos, típicamente está seguida por la recuperación del estado previo al tratamiento.
NAPROXENO y sus metabolitos se eliminan principalmente por los riñones; por tanto, el fármaco debe ser usado con precaución en pacientes con insuficiencia renal importante, y en estos pacientes se recomienda la vigilancia de creatinina en suero y/o la depuración de creatinina.
También se debe tener precaución si el fármaco es administrado a pacientes con depuración de creatinina menor de 20 ml/minuto, ya que se ha visto acumulación de metabolitos de NAPROXENO en estos pacientes.
La enfermedad hepática alcohólica crónica y probablemente otras enfermedades con niveles bajos o anormales de proteínas plasmáticas (albúmina) reducen la concentración plasmática total de NAPROXENO, pero la concentración plasmática de NAPROXENO libre está aumentada.
Se debe recomendar precaución cuando se requieran dosis altas, y se puede requerir cierto ajuste de la dosis en estos pacientes. Es prudente usar la dosis efectiva más baja.
Los estudios en ancianos indican que aunque la concentración plasmática total de NAPROXENO no cambia, aumenta la fracción libre de NAPROXENO en plasma. Se debe recomendar precaución cuando se requieran dosis altas, y se puede requerir cierto ajuste de la dosis en los pacientes ancianos. Al igual que con otros fármacos usados en los ancianos, es prudente usar la dosis efectiva más baja.
Función hepática: Al igual que con otros agentes antiinflamatorios no esteroideos, hasta en 15% de los pacientes pueden ocurrir aumentos hacia el límite superior de una o más pruebas hepáticas. Estas anormalidades pueden progresar, permanecer esencialmente sin cambio, o ser pasajeras durante la terapia continua. Probablemente la prueba de TGP (ALT) es el indicador más sensible de trastorno hepático. En estudios clínicos controlados ocurrieron aumentos significativos (tres veces el límite superior normal) de TGP o TGO en menos del 1% de los pacientes. Un paciente con síntomas y/o signos que sugieren trastorno hepático, o en quien ha ocurrido un resultado anormal en una prueba hepática, debe ser evaluado para detectar evidencia de desarrollo de una reacción hepática más severa mientras está recibiendo terapia con NAPROXENO.
Las reacciones hepáticas severas, incluyendo ictericia y casos fatales de hepatitis, han sido reportadas con NAPROXENO al igual que con otros agentes antiinflamatorios no esteroideos.
Aunque son reacciones raras, NAPROXENO debe ser interrumpido si los resultados anormales de las pruebas hepáticas persisten o empeoran, si aparecen signos y síntomas clínicos consistentes con enfermedad hepática, o si ocurren manifestaciones sistémicas (por ejemplo, eosinofilia y erupción).
Retención de líquido y edema: Se ha observado edema periférico en algunos pacientes tratados con NAPROXENO. Como cada tableta de NAPROXENO sódico contiene 25 ó 50 mg de sodio (aproximadamente 1 mEq por cada 250 mg de NAPROXENO), esto se debe considerar en pacientes cuyo consumo total de sodio debe ser restringido grandemente. Por estas razones, NAPROXENO sódico en tabletas debe usarse con precaución en pacientes con retención de líquidos, hipertensión o insuficiencia cardiaca.
Las siguientes reacciones adversas están divididas en tres grupos, con base en la posibilidad de una relación causal entre NAPROXENO y estas acciones adversas. Para las reacciones en el grupo de probable relación causal existe por lo menos un caso para cada reacción adversa, donde hay evidencia que sugiere la existencia de una relación causal entre el uso del fármaco y el evento reportado.
En general, las reacciones adversas en pacientes tratados crónicamente fueron reportadas 2 a 10 veces más frecuentemente que en los estudios a corto plazo en 962 pacientes tratados a causa de dolor leve a moderado o dismenorrea. Los malestares más frecuentemente reportados se refieren al tracto gastrointestinal. Un estudio clínico encontró que en casos de artritis reumatoidea, las reacciones gastrointestinales son más frecuentes y más severas en los pacientes que toman dosis diarias de 1,500 mg de NAPROXENO en comparación con los pacientes que toman 750 mg de NAPROXENO.
En estudios clínicos controlados aproximadamente 80 niños, y en estudios abiertos bien vigilados cerca de 400 niños con artritis juvenil, tratados con NAPROXENO, la incidencia de erupción y los tiempos prolongados de sangrado estuvieron aumentados, la incidencia de reacciones en el sistema gastrointestinal y nervioso central fueron casi las mismas, y la incidencia de otras reacciones fue menor en los niños que en los adultos. Las siguientes reacciones adversas se dividen en tres grupos, con base en la frecuencia y relación causal.
Incidencia mayor de 1% (probable relación causal):
Gastrointestinales: Constipación, acidez, dolor abdominal, náusea, dispepsia, diarrea y estomatitis.
Sistema nervioso central: Cefalea, vértigo, somnolencia y mareo.
Dermatológicas: Prurito, erupciones cutáneas, equimosis, sudoración y púrpura.
Sentidos especiales: Tinnitus, trastornos auditivos y trastornos visuales.
Cardiovasculares: Edema, disnea y palpitaciones.
Generales: Sed.
Incidencia menor del 1% (probable relación causal): Las siguientes reacciones adversas fueron reportadas con una frecuencia menor de 1% durante estudios clínicos controlados y mediante reportes de voluntarios desde la comercialización del NAPROXENO:
Gastrointestinales: Pruebas anormales de función hepática, colitis, sangrado gastrointestinal y/o perforación, hematemesis, ictericia, pancreatitis, melena y vómito.
Renales: Nefritis glomerular, hematuria, hiperpotasemia, nefritis intersticial, síndrome nefrótico, enfermedad renal, insuficiencia renal y necrosis papilar renal.
Hematológicos: Agranulocitosis, eosinofilia, granulocitopenia, leucopenia y trombocitopenia.
Sistema nervioso central: Depresión, anormalidades en el sueño, incapacidad para concentrarse, insomnio, malestar, mialgia y debilidad muscular.
Dermatológicas: Alopecia, dermatitis fotosensitiva, urticaria, rashes cutáneos, reacciones de fotosensibilidad que simulan porfiria cutánea tardía y epidermólisis ampollosa.
Sentidos especiales: Trastornos auditivos.
Cardiovasculares: Insuficiencia cardiaca congestiva.
Respiratorias: Neumonitis eosinofílica.
Generales: Reacciones anafilactoides, edema angioneurótico, trastornos menstruales y pirexia (escalofríos y fiebre).
Incidencia menor del 1% (relación causal desconocida): Estas observaciones se indican para que sirvan como información de alerta para el médico.
Hematológicas: Anemia aplásica y anemia hemolítica.
Sistema nervioso central: Meningitis aséptica y trastornos cognoscitivos.
Dermatológicas: Necrólisis epidérmica, eritema multiforme y síndrome de
Stevens-Johnson.
Gastrointestinales: Ulceración gastrointestinal no péptica y estomatitis ulcerativa.
Cardiovasculares: Vasculitis.
Generales: Hiperglucemia e hipoglucemia.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: El uso de AINE en pacientes que reciben inhibidores de la ECA puede potenciar los estados de enfermedad renal.
Los estudios realizados in vitro han demostrado que el anión NAPROXENO, dada su afinidad por la proteína, puede desplazar de sus sitios de unión a otros fármacos que también se unen a la albúmina.
Teóricamente, a su vez, el anión NAPROXENO también puede ser desplazado por otros fármacos. Estudios controlados a corto plazo no pudieron demostrar que la administración del fármaco afecte significativamente los tiempos de protrombina cuando es administrado a individuos que toman anticoagulantes tipo cumarina. Sin embargo, se debe tener precaución, ya que se han visto interacciones con otros agentes no esteroideos de esta clase. Similarmente, los pacientes que toman el fármaco y una hidantoína, sulfonamida o sulfonilurea, deben ser observados para detectar signos de toxicidad causada por estos fármacos.
No se recomienda la administración concomitante de NAPROXENO y ácido acetilsalicílico porque NAPROXENO es desplazado de sus sitios de unión, resultando en menores concentraciones plasmáticas y menores niveles plasmáticos máximos.
Se ha reportado que el efecto natriurético de la furosemida es inhibido por algunos fármaco de esta clase. También se ha reportado inhibición de la depuración renal del litio, con aumentos en las concentraciones plasmáticas de litio.
NAPROXENO y otros agentes antiinflamatorios no esteroideos pueden reducir el efecto antihipertensivo del propranolol y otros bloqueadores beta. Se debe tener precaución si NAPROXENO es administrado de manera concomitante con metotrexato, se ha reportado que NAPROXENO, NAPROXENO sódico y otros fármacos antiinflamatorios no esteroideos reducen la secreción tubular de metotrexato en un modelo en animales, aumentando posiblemente la toxicidad del metotrexato.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Se realizó un estudio de dos años en ratas para evaluar la posible carcinogenicidad del NAPROXENO, usando dosis de 8, 16 y 24 mg/kg/día (50, 100 y 150 mg/m2).
La dosis máxima usada fue 0.28 veces la exposición sistémica en humanos a la dosis recomendada. No se encontró evidencia de carcinogénesis ni teratogénesis.
En los estudios en animales de experimentación no se observaron trastornos de la fertilidad ni daño fetal.
No se han realizado estudios adecuados y bien controlados en mujeres embarazadas. Como los estudios de reproducción animal no siempre predicen la respuesta en humanos, NAPROXENO no debe ser usado durante el embarazo, a menos que sea absolutamente necesario.
La acción principal de NAPROXENO sódico sobre la inhibición de las prostaglandinas posiblemente sea el mecanismo por el cual el trabajo de parto se prolongue y cause distocia, fenómeno observado en animales de experimentación.
Aunque esto no ha sido investigado en el humano, un hecho es que NAPROXENO sódico facilita como sucede con otros fármacos de la misma clase, la oclusión temprana del conducto arteriovenoso del producto de la gestación. Por tales condiciones, NAPROXENO no deberá utilizarse durante el embarazo.
NAPROXENO es excretado por la leche materna, por lo que en madres que tengan planeada la alimentación al seno materno, debe evitarse la prescripción de NAPROXENO sódico.
No se han realizado estudios en niños menores de 2 años, por tanto, su prescripción debe evitarse.
DOSIS Y VÍA DE ADMINISTRACIÓN:
En la población pediátrica, la dosificación es la siguiente:
Dosis de inicio de 10 mg/kg seguida por 2.5 a 5 mg/kg cada 8 horas. La dosis no deberá exceder de 15 mg/kg al día después del primer día de tratamiento.
En caso de artritis reumatoide juvenil, la dosis usual es de 10 mg/kg diariamente, dividida en dos tomas con intervalos de 12 horas, junto con los alimentos.
Para artritis reumatoidea, osteoartritis y espondilitis anquilosante la terapia inicial es de 1,000 mg al día, en dos tomas o en dosis única.
Durante la administración prolongada, la dosis de NAPROXENO se puede aumentar o disminuir, dependiendo de la respuesta clínica del paciente. Una dosis diaria menor puede ser suficiente para la administración prolongada.
Las dosis en la mañana y en la noche no tienen que ser de la misma magnitud, y no es necesaria la administración del fármaco más de dos veces al día.
En pacientes que toleran bien dosis menores, la dosis se puede aumentar hasta 1,500 mg de NAPROXENO por día durante periodos limitados, cuando se requiere un mayor nivel de actividad antiinflamatoria/analgésica. Cuando los pacientes son tratados con 1,500 mg/kg de NAPROXENO, el médico debe observar beneficios clínicos suficientes que superen el posible riesgo aumentado.
Artritis juvenil: La dosis diaria total recomendada de NAPROXENO es aproximadamente 10 mg/kg, administrada en 2 dosis divididas (es decir, 5 mg/kg dos veces al día).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Una dosis excesiva del fármaco puede caracterizarse por somnolencia, pirosis, indigestión, náuseas, vómito, hipoprotrombinemia, disfunción renal, acidosis metabólica, apnea, desorientación. Algunos pacientes experimentan convulsiones, no siendo clara la relación con NAPROXENO podría ser letal.
Si un paciente ingiere una cantidad excesiva de NAPROXENO a propósito, o accidentalmente, se recomienda un lavado gástrico y las medidas usuales de soporte.
Los estudios en animales indican que la rápida administración de una cantidad adecuada de carbón activado puede reducir de manera significativa la absorción del fármaco, debido a su alta afinidad a las proteínas plasmáticas.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a temperatura ambiente a no más de 30° C y en lugar seco.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se
deje al alcance de los niños.
No se administre durante el embarazo ni en la lactancia.
NOMBRE Y DOMICILIO DEL LABORATORIO:
Véase Presentación o Presentaciones.
PRESENTACIÓN O PRESENTACIONES:
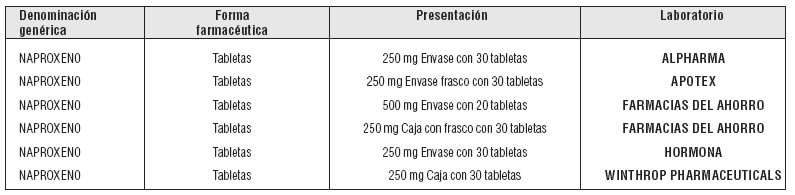
Fuente: S.S.A. Catálogo de Medicamentos Genéricos Intercambiables para
farmacias y público en general al 3 de agosto de 2007.
Con el objeto de demostrar la intercambiabilidad a que se refiere el artículo
75 del reglamento de Insumos para la Salud, los medicamentos que integran
el Catálogo de Medicamentos Genéricos Intercambiables han sido comparados,
siguiendo los lineamientos indicados por la NOM-177SSA1-1998,
contra los productos innovadores o de referencia enlistados en las págs. 11 a
22 donde usted lo podrá consultar.